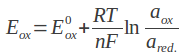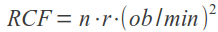Wymownym znakiem obecnych czasów jest możliwość stworzenia wartościowej publikacji naukowej bez wychodzenia z domu. Ba, nawet bez wstawania od komputera. Przemierzając serwery wypełnione sekwencjami całych genomów bakteryjnych oraz dysponując odpowiednimi narzędziami bioinformatycznymi, jesteśmy w stanie wyłuskać więcej informacji na temat poszczególnych mikroorganizmów, niż było to możliwe kiedykolwiek wcześniej. W dobie sekwencjonowania nowej generacji osiągnęliśmy bowiem nie tylko niesłychany wzrost przepustowości i dokładności sekwencjonowania, ale byliśmy także w stanie przekroczyć granice tego, co niehodowalne w warunkach laboratoryjnych.
10 lat pangenomu
W miarę wzrostu liczby sekwencji genomowych poszczególnych szczepów nie dało się nie zauważyć olbrzymiego dysonansu pomiędzy sekwencjami przedstawicieli danego gatunku. Wśród nich można było wskazać zbiór genów wspólnych oraz tych odróżniających od siebie poszczególne szczepy. Pojęcie pangenomu po raz pierwszy wprowadził Tettelin i współpracownicy w 2005 r. Analizując sekwencje ośmiu przedstawicieli paciorkowca Streptococcus agalactiae, zadali pytanie o ilość sekwencji genomowych potrzebnych do pełnego opisu danego gatunku. Porównując otrzymane sekwencje z istniejącymi w bazach danych, autorzy zaproponowali definicję pangenomu złożonego z genomu rdzeniowego – a więc zestawu genów wspólnego dla wszystkich szczepów (około 80%) - oraz genomu pomocniczego, zawierającego geny częściowo wspólne i charakterystyczne dla poszczególnych szczepów. Pangenom, czyli pulę wszystkich genów danego gatunku, określić można jako otwarty (nieskończony) lub zamknięty (skończony). Pangenom zamknięty jest charakterystyczny dla gatunków zasiedlających wąskie nisze ekologiczne lub bezwzględnych pasożytów, które nie są poddane presji selekcyjnej. Przykładem bakterii o skończonym pangenomie jest wąglik Bacillus anthracis. Wielokrotnie uzyskiwane sekwencje genomowe tego gatunku cechują się stałą wielkością genomu pomocniczego. Pangenom otwarty jest charakterystyczny dla gatunków, które na drodze przystosowania ewolucyjnego wykształciły zdolność do pobierania obcego materiału genetycznego ze środowiska na drodze horyzontalnego transferu genów (a więc w procesie transformacji, transfekcji z udziałem bakteriofagów lub koniugacji). W skład ich genomu pomocniczego wchodzą zatem charakterystyczne struktury, takie jak wyspy patogeniczności i lekooporności, sekwencje insercyjne i transpozony oraz plazmidy. W przypadku pangenomu otwartego, zależność przedstawiająca ilość nowych genów względem ilości dostępnych genomów jest malejącą funkcją asymptotyczną. Według dostępnych modeli matematycznych, nowe geny takiego gatunku będą pojawiać się nawet po uzyskaniu sekwencji setek kolejnych genomów. Pangenomem otwartym cechują się takie patogeny człowieka jak Streptococcus agalactiae, Staphylococcus aureus, Escherichia coli czy Enterococci.
 Na podstawie Rouli et al. 2015
Na podstawie Rouli et al. 2015
Od przybytku głowa nie boli?
Dokonująca się zmiana w koncepcji genomu stawia zasadnicze pytanie o taksonomię mikroorganizmów. W obliczu tak wielkiej różnorodności sekwencji, jaka wielkość genomu rdzeniowego decyduje o przynależności do tego samego gatunku? Czy w tej perspektywie genotypowanie na podstawie jednego genu, tj. sekwencji 16S rRNA, wciąż jest zasadne? Porównanie dużej ilości sekwencji pokazuje, że faktyczne zróżnicowanie genomów nie znajduje odzwierciedlenia w grupowaniu serologicznym czy typach MLST (ang. multilocus sequence typing). Braku tej korelacji należy się doszukiwać w dystrybucji badanych genów w obrębie genomu rdzeniowego i pomocniczego – antygeny otoczkowe zazwyczaj kodowane są w genomie pomocniczym, zaś geny wykorzystywane w grupowaniu MLST należą do genomu rdzeniowego. Dyskryminacja gatunków na poziomie sekwencji genomowej poddaje także w wątpliwość rozróżnianie gatunków z rodzaju Shigella. Zarówno homologia sekwencji 16S rRNA, jak i uzyskane w ostatnim czasie sekwencje genomowe wskazują na większe podobieństwo między wybranymi szczepami Shigella spp. i E. coli niż między szczepami w obrębie tych gatunków. W świetle dzisiejszej wiedzy, wyodrębnienie rodzaju Shigella z gatunku E. colijest więc oparte wyłącznie o kryteria fenotypowe.
Okres fascynacji wyodrębnionymi mechanizmami zjawisk biologicznych w tak zwanej filozofii redukcjonizmu wydaje się być dogasającym rozdziałem współczesnej nauki. Święcące swoje triumfy podejście holistyczne stara się udowodnić, że genetyki populacji nie da się zredukować do biologii molekularnej. Analizy porównawcze dużych ilości danych pochodzących z różnych szczepów pozwalają lepiej zrozumieć różnorodność biologiczną bakterii i przestają być jedynie teoretycznym ćwiczeniem. Kto wie, ile niespodzianek kryją jeszcze sekwencje mikroorganizmów żyjących wokół nas.
Monika Kossakowska-Zwierucho